Tercer Milenio
En colaboración con ITA
Progeria: un viaje hacia la esperanza y la curación
La progeria es una enfermedad genética extremadamente rara y fatal que causa un envejecimiento prematuro y acelerado.


La búsqueda de la inmortalidad ha sido un tema recurrente a lo largo de la historia de la humanidad. Aunque su búsqueda impulsa el progreso científico y tecnológico, también plantea desafíos éticos y morales que requieren una reflexión cuidadosa entre el deseo humano y el respeto por los límites naturales de la existencia. La longevidad excepcional de Jeanne Calment (122 años) la convirtió en un fenómeno científico y cultural. Entre 2020 y 2050, se espera que el número de personas que superen los 80 años se triplique. En contraste, los niños con progeria generalmente no superan la adolescencia.
Crecer y envejecer a la vez
En octubre de 1996 un niño llamado Sam comenzó su viaje en este mundo. Parecía estar sano al nacer, pero al cabo de un año sus padres sospecharon que algo no estaba bien. Fue diagnosticado a los 22 meses de un síndrome conocido como progeria, una enfermedad genética extremadamente rara y fatal que causa un envejecimiento prematuro y acelerado; con una esperanza de vida no más allá de la adolescencia debido a complicaciones vasculares.
A los pocos meses empezó a desarrollar la apariencia física que caracteriza a estos niños. Sam comenzó a percibir que el tiempo transcurría para él más rápido que para otros niños, mientras observa cómo su cuerpo infantil envejece con la inocencia y la asombrosa percepción de un niño.
Fue entonces cuando sus padres decidieron actuar. Ante la falta de información científica sobre la progeria y con el deseo de encontrar respuestas y soluciones para su hijo y otros niños afectados, decidieron fundar una organización dedicada a la investigación de esta enfermedad. En 1999, crearon una fundación con el objetivo de recaudar fondos y reclutar investigadores comprometidos con el estudio de la progeria, así nació la Progeria Research Foundation.
Esta fundación no solo proporciona recursos financieros para la investigación, sino que también desempeña un papel crucial en la creación de conciencia sobre la progeria, en el asesoramiento de las familias y en la conexión de personas interesadas en colaborar en la búsqueda de tratamientos y terapias efectivas.


Una proteína anómala
Pronto llegaron los frutos. El primer hito ocurrió en 2003, cuando se identificó la causa molecular de la enfermedad: una mutación genética que produce una proteína anómala llamada progerina. Este descubrimiento representó un punto de inflexión en la comprensión de esta dolencia. A partir de entonces, fue posible realizar un diagnóstico genético de la enfermedad, brindando a los padres de niños afectados una mayor claridad y certeza sobre la condición de sus hijos. Por otra parte, se allanó el camino para futuras investigaciones y el desarrollo de enfoques terapéuticos más efectivos para la progeria.
La generación de un ratón modificado genéticamente por el equipo liderado por el aragonés Carlos López Otín nos ha posibilitado el estudio de la enfermedad a un nivel más fundamental, lo que a su vez ha facilitado la búsqueda de terapias que puedan mejorar la calidad de vida de estos niños.
La creación de modelos experimentales representa un segundo hito crucial, ya que proporciona una plataforma para avanzar en la investigación contra la progeria. Estos modelos permiten validar la efectividad de tratamientos, incluyendo los dirigidos a reducir la cantidad de progerina, la proteína anómala que también se acumula en adultos sanos durante el envejecimiento normal, aunque en niveles mucho más bajos que en niños con progeria.
El primer tratamiento
Así, y después de muchos años de investigación y esfuerzo, llegó el tercer gran hito de este viaje: la aprobación del primer tratamiento contra esta dolencia (2020), que consigue prolongar una media de dos años y medio la longevidad de los niños con progeria.
La cura definitiva de la enfermedad está aún por llegar, aunque ya se han hecho importantes avances al respecto. En concreto, los grupos dirigidos por los españoles Juan Carlos Ispizúa Belmonte y Carlos López Otín demostraron, por primera vez, las ventajas de la terapia génica en ratones con progeria. Si bien es cierto que todavía hay que mejorar la tecnología y se necesita un debate profundo sobre los aspectos éticos y morales de su aplicación, tarde o temprano esta posibilidad terapéutica será factible.

Mientras llega la cura definitiva, y con ello el último gran hito de esta historia, las investigaciones que realizamos en diversos grupos, se centran en aliviar las afecciones asociadas a la progeria, mejorando así la calidad de vida de estos niños.
La iniciativa de los padres de Sam (recogida en el documental ‘La vida según Sam’, disponible en HBO y Prime Video) es realmente inspiradora y ejemplifica cómo el apoyo a la investigación, de forma privada o pública, puede tener un impacto positivo en la sociedad. Desde el momento en que nació, Sam comenzó su propio viaje lleno de aventuras, desafíos y momentos especiales. Con el tiempo, acumuló un tesoro de experiencias que moldearon su personalidad y su visión del mundo.
Causa, síntomas y tratamiento
La progeria, específicamente el síndrome de Hutchinson-Gilford, es causada por una mutación genética específica en el gen LMNA (lamin A/C). Este gen proporciona instrucciones para producir las proteínas lamin A y lamin C (laminas), que son componentes estructurales importantes de la envoltura nuclear de las células. La mutación genética asociada con la progeria conduce a la producción anormal de una forma de lamin A llamada progerina, que se acumula en el núcleo de las células y afecta a su estructura y función. Esto conduce a una serie de cambios celulares y moleculares que dan como resultado el envejecimiento prematuro de los individuos afectados.
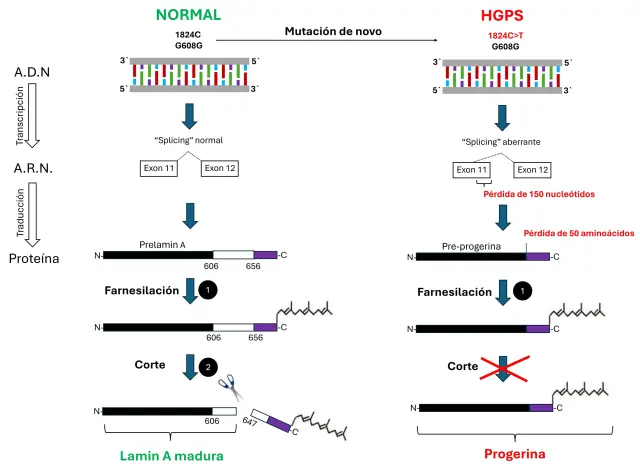
La progeria es un trastorno genético extremadamente raro y generalmente se produce de manera esporádica, lo que significa que no suele ser hereditario. La mayoría de los casos son causados por una nueva mutación genética que ocurre durante la formación de los gametos (espermatozoides u óvulos) o en las primeras etapas del desarrollo embrionario.
Los niños con progeria muestran características de envejecimiento acelerado, incluyendo problemas de salud relacionados con el envejecimiento. Aunque los síntomas pueden variar de un niño a otro, a menudo tienen una apariencia facial distintiva, incluyendo cabeza pequeña, mandíbula subdesarrollada, nariz estrecha y prominente, piel delgada y arrugada, y pérdida de cabello.
También pueden tener un crecimiento deficiente, lo que resulta en baja estatura y peso por debajo del promedio para su edad. La rigidez articular es común en la progeria, lo que puede dificultar el movimiento y limitar la movilidad de los niños afectados. Los niños con progeria pueden experimentar una variedad de problemas de salud que incluyen pérdida de grasa corporal y tejido subcutáneo, diabetes, problemas dentales y óseos, pérdida de audición y problemas de visión. Además, la progeria está asociada con un mayor riesgo de enfermedades cardiovasculares, como arteriosclerosis (endurecimiento de las arterias), enfermedades del corazón y accidentes cerebrovasculares.
El tratamiento de la progeria es un desafío debido a la naturaleza compleja y poco común de esta enfermedad genética. Los pacientes con progeria suelen requerir una atención médica integral y coordinada para abordar todas las dimensiones de su salud, incluidos los aspectos físicos, emocionales y sociales. Aunque todavía no existe una cura definitiva para la progeria (terapia génica), se han desarrollado tratamientos que se centran en aliviar los síntomas y mejorar la calidad de vida de los niños.
El lonafarnib es un medicamento prometedor en el tratamiento de la progeria, ya que ha demostrado ser eficaz en la reducción de los niveles de progerina. El lonafarnib pertenece a una clase de fármacos conocidos como inhibidores de la farnesiltransferasa, que actúan bloqueando una enzima llamada farnesiltransferasa, involucrada en la producción de la proteína progerina. En 2020, la Administración de Alimentos y Medicamentos de Estados Unidos (FDA) aprobó el lonafarnib como el primer tratamiento específico para la progeria, lo que representó un avance significativo en el manejo de esta enfermedad.
Valiosas lecciones biológicas: ¿por qué envejecemos?
El envejecimiento es un proceso complejo y multifacético que afecta a todos los organismos vivos.
Aunque no hay una causa única, se han identificado al menos nueve aspectos distintivos que ofrecen una visión completa de los procesos subyacentes y sus efectos en la salud y la longevidad. Incluyen: mutaciones y daños en el ADN (inestabilidad genómica); cambios en la expresión génica; pérdida de la capacidad para mantener correctamente las proteínas (proteostasis); deterioro en la función de las mitocondrias, las estructuras celulares responsables de la producción de energía; senescencia celular o detención irreversible del crecimiento celular; reducción de la longitud de los extremos de los cromosomas (telómeros); disminución en la capacidad de autorrenovación y diferenciación de las células madre, lo que limita la capacidad de reparación y regeneración de los tejidos; alteración en la detección de los nutrientes; y alteraciones en la comunicación entre las células y los tejidos.
La progeria proporciona valiosas lecciones sobre el envejecimiento y ofrece información única sobre los procesos biológicos implicados en este fenómeno. Esta enfermedad está relacionada con mutaciones en el gen LMNA, que codifica una proteína esencial para mantener la integridad del núcleo celular. Esto resalta la importancia de un núcleo celular funcional en la regulación del envejecimiento y la salud celular en general.
Los niños con progeria tienen un alto riesgo de desarrollar enfermedades cardiovasculares, como enfermedades del corazón y accidentes cerebrovasculares, que son comunes en el envejecimiento normal y las principales causas de muerte en países desarrollados. Esto subraya la estrecha relación entre el envejecimiento y las enfermedades cardiovasculares y destaca la importancia de mantener la salud cardiovascular para promover un envejecimiento saludable.
En cifras
Desde su fundación, la Progeria Research Foundation (PRF) ha recaudado, mediante donaciones, más de 90 millones de dólares. Con este dinero se han podido financiar cinco ensayos clínicos de medicamentos contra la progeria, 85 proyectos de investigación y 14 reuniones científicas internacionales sobre progeria. Más del 80% de los gastos anuales de PRF se dedican a sus programas y materiales de atención médica, que están traducidos a 38 idiomas.
En el base de datos médica y de investigación de la PRF, hay 221 niños. Hoy en día 196 niños, de 50 países, viven con algún tipo de laminopatías progeroides; de ellos, 144 tienen el síndrome de Hutchinson-Gilford.
Las laminopatías progeroides son un grupo de trastornos genéticos raros que afectan a las proteínas laminas, componentes estructurales del núcleo celular. Estos trastornos presentan síntomas similares a la progeria. Sin embargo, las laminopatías progeroides engloban un espectro más amplio de trastornos que pueden afectar a personas de diferentes edades y con diferentes grados de gravedad. Ejemplos de laminopatías progeroides incluyen el síndrome de Hutchinson-Gilford (progeria clásica), el síndrome de Werner y la lipodistrofia familiar parcial. El tratamiento se centra en el manejo de los síntomas y el apoyo integral al paciente y su familia, ya que no hay una cura específica para estos trastornos.
Ricardo Villa Bellosta Centro Singular de Investigación en Medicina Molecular y Enfermedades Crónicas de la Universidad de Santiago de Compostela
-Ir al suplemento Tercer Milenio
Apúntate y recibe cada semana en tu correo la newsletter de ciencia